Ведущую роль в этом процессе отводят наследственности – способности передавать генетический материал от родителей к потомкам.


Чаще всего всё проходит безошибочно, но иногда при наследовании информации случаются дефекты. В результате возникают выкидыши, мертворождение или рождение детей с генетическими аномалиями. На данный момент известно более 1000 аномалий, а более 100 из них подробно описаны и изучены.
Важно: примерно десятая часть яйцеклеток имеет различные хромосомные аномалии. В таком случае чаще всего на ранних неделях внутриутробного развития происходит самопроизвольное прерывание беременности.
Что такое ген и хромосома

Ген – это материальный носитель определённого наследственного признака. Он кодирует один белок. Он состоит из дезоксирибонуклеиновой кислоты – ДНК. Молекула ДНК – длинная спираль с поперечными перемычками. Она очень похожа на закрученную вокруг своей оси лестницу. Каждая ступень состоит из 4 типов молекул, которые называют нуклеотидами: аденин, тимин, гуанин и цитозин. Каждый нуклеотид имеет свою пару, с которой соединяется: аденин – с тимином, а гуанин – с цитозином. То есть гуанин не может образовать пару с тимином.
ДНК содержит специальный код, который несёт информацию о синтезе белка. Размеры генов, как и размеры белков, разные. Именно белок является основной структурой организма. Он выполняет множество функций, одна из которых – это ферменты.
Ферменты – это сложные белки, которые приводят в действие множество процессов в организме. Они выступают в роли катализаторов химических реакций, помогают трансформировать одни вещества в другие. На данный момент известно более 5000 ферментов.
Вся структура и функционирование организма управляются белковыми структурами. Синтез белков контролируется генами внутри хромосомы. Хромосомы – это длинная нить ДНК, содержащая множество генов. Картина полного набора хромосом в ядре называется кариотипом, а процесс его расшифровки – кариотипированием.
В одной хромосоме содержится от сотен до тысяч генов. Всего у человека насчитывается примерно 22000–23000 генов. Они распределены по хромосомам в строгом порядке.
Важно: в норме каждая клетка человека содержит одинаковое количество хромосом: 46 штук или 23 пары. Исключение – половые клетки. Они содержат ровно половину. Это нужно для того, чтобы при слиянии сперматозоида с яйцеклеткой получилось как раз 23 пары – набор нормального человека.
Индивидуальная комбинация генов человека определяет его генотип. Генотип – это полный набор генов, унаследованный от обоих родителей. Иными словами, это набор программ, в которых прописано, какие белки и в каком количестве будут синтезироваться у человека.
Фенотип – это проявление генотипа. То есть визуальное строение человека: цвет глаз, волос, тембр голоса и многое другое. Фенотип может меняться при разных условиях окружающей среды. Например, у альпинистов от постоянного воздействия стираются линии на подушечках пальцев. Это изменения фенотипа, но не генотипа. У детей альпинистов с линиями на пальцах будет всё в порядке.
Как образуется «поломка» в геноме
«Поломка» в геноме называется мутацией. Это изменение одного или нескольких генов. Чаще всего мутации вредны, но есть и исключения. Например, полоски у зебр – это следствие мутации гена, который отвечает за окраску животного. В результате одна его часть была заменена другой. В итоге на свет появилась уникальная полосатая зебра.
Мутации бывают спонтанными и индуцированными. Спонтанные возникают случайно. Пример спонтанной мутации – разноцветные котята в помёте. Причины возникновения спонтанных мутаций до сих пор окончательно не ясны.
Индуцированные мутации возникают под воздействием каких-либо внешних факторов. Это могут быть радиоактивное излучение, вирусы, химические вещества, лекарства и многое другое. Индуцированные мутации чаще наследуются и могут наносить вред организму. По степени поражения выделяют 4 вида мутаций.
Генные возникают при замещении одного гена другим, выпадении или добавлении одного гена. Примером генной мутации служат болезни, где нарушен синтез только одного белка: фенилкетонурия или альбинизм.
Хромосомные возникают при изменении структуры хромосом. Чаще всего это происходит при нарушении деления клеток. Известно 5 патологических процессов:
- утрата – потеря хромосомой своей хвостовой части,
- делеция – потеря среднего участка хромосомы,
- дупликация – удвоение участка хромосомы,
- инверсия – поворот участка хромосомы на 180 градусов,
- транслокация – перенос участка одной хромосомы на другую.
При таких аномалиях возникает нарушение сразу нескольких процессов, что ведёт к значительному урону для человеческого организма. Например, синдром Прадера – Вилли: ожирение, низкий рост и умственная отсталость.
Геномные мутации поражают сразу весь геном. Обычно это происходит при нарушении расхождения хромосом. Изменение количества хромосом в половой клетке может быть кратным (2n, 3n и так далее). Это называется полиплоидией. Некратное изменение (n + 1, n + 2) называют анеуплоидией. Самый известный геномный синдром – синдром Дауна, когда не расходится 21 пара хромосом. Ребёнок получает не две 21 хромосомы, а три.
Цитоплазматические мутации связаны с мутацией генов, которые находятся вне ядер. У клеток человека собственное ДНК есть у митохондрии, «силовой станции» клетки. Данный вид мутации изучен слабее всего.
Виды наследования
Мутации, из-за которых возникают наследственные болезни, могут иметь доминантный или рецессивный характер наследования. При аутосомно-доминантном наследовании болезнь проявляется, если у человека есть хотя бы один мутированный ген. При аутосомном наследовании за развитие признаков отвечают гены, расположенные не на половых Х и Y-хромосомах. Таким образом передаются некоторые виды рака молочной железы и синдром Марфана.
Аутосомно-рецессивное наследование – это вид, при котором болезнь проявляется в том случае, если дефектный ген был унаследован от обоих родителей. И при этом не содержится в половых Х и Y-хромосомах.
Человек, имеющий только одну копию дефектного гена, чаще всего является почти или полностью здоровым. Это называется «носитель болезни». Если его партнёр тоже окажется носителем такого же дефектного гена, то с вероятностью 25% их ребёнок унаследует оба гена. И родится больным. Таким образом наследуют фенилкетонурию и спинальную мышечную атрофию.
Ещё один вид наследования – Х-сцепленное рецессивное. Дефектный ген находится на Х-хромосоме. Болезнь проявляется тогда, когда второй Х-хромосомы с нормальной копией этого же гена у человека нет. Это возможно при кариотипе XY или передаче второй больной хромосомы при кариотипе XX.
Например, отец с рецессивным и связанным с X-хромосомой заболеванием передаст свою здоровую Y-хромосому сыновьям. Все они будут здоровы. Если он передаст свою Х-хромосому с дефектным геном дочерям, то они будут носителями болезни.
Женщины, имеющие только один дефектный ген в одной хромосоме, передадут болезнь примерно половине своих детей. Если у женщины дефектный ген в обоих X-хромосомах, то его получат все дети. По такому типу наследуется гемофилия и дальтонизм.
Методы диагностики
Впервые на такие болезни стали обширно исследовать в 60-х годах прошлого века. Именно в это время возникли мощные микроскопы и множество научных разработок, которые позволили глубже разобраться в структурах генов человека. На данный момент есть несколько современных способов расшифровки генов.
Кариотипирование
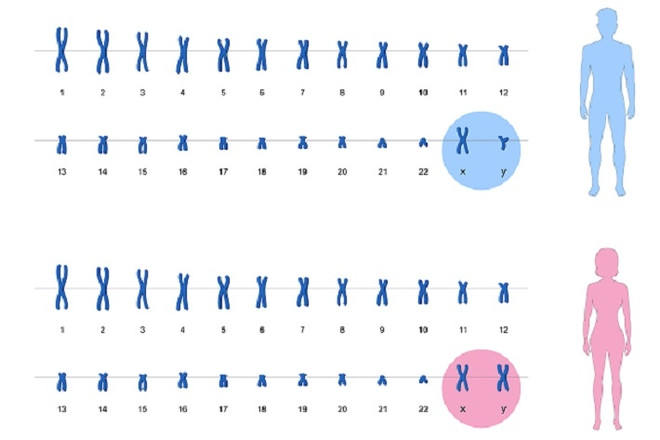
Исследование хромосомного набора человека. Позволяет определить количество и вид хромосом. Нормальный кариотип женщины – 46 XX, мужчины – 46 XY.
Х и Y – это половые хромосомы, именно они определяют пол будущего ребёнка. От мамы ребёнок может получить только X-хромосому, а от папы – и X, и Y. То есть за пол ребёнка отвечает только мужчина.
Чаще всего к кариотипированию прибегают супружеские пары, у которых возникли проблемы с деторождением:
- при привычном невынашивании беременности;
- при бесплодии неясной этиологии;
- при повторных диагнозах «неразвивающаяся беременность», «анэмбриония» или «пузырный занос»;
- при неудачных попытках ЭКО;
- при мертворождении предыдущего ребёнка;
- при рождении детей с хромосомной патологией или с множественными пороками развития.
Также к кариотипированию прибегают для определения пола ребёнка. При некоторых патологиях наружные половые органы развиваются неправильно. В этом случае половую принадлежность бывает сложно определить «на глаз». Иногда кариотипирование необходимо при нарушении развития ребёнка или умственной отсталости. Это позволит понять, чем человек болен и есть ли надежда на излечение.
FISH - Флуоресцентная гибридизация (fluorescent in situ hybridization)
Кариотипирование позволяет определить только количество и последовательность хромосом. Метод FISH выявляет последовательности ДНК на хромосомах. Является более подробным, чем кариотипирование. Но при одном исследовании можно определить только одно нарушение гена. То есть этим методом стоит пользоваться, когда уже есть подозрение на патологию определённого участка. Это нужно, чтобы подтвердить или опровергнуть это предположение. Метод не покажет все сломанные участки.
Хромосомный микроматричный анализ
При этом исследовании фиксируются все патологические участки генома. Этот метод используют как более расширенную и углублённую замену кариотипированию. Часто проводят у пациентов с умственной отсталостью, множественными пороками развития.
Самые известные болезни хромосомной природы

Самым известным хромосомным заболеванием является синдром Дауна. Впервые эта болезнь была описана английским врачом Джоном Лэнгдоном Дауном в 1866 году. Но только в 1959 году французский учёный Жером Лежен доказал его генетическую природу. Ранее синдром Дауна называли монголоидной идиопатией. И лишь в 20 веке болезнь получила имя своего первооткрывателя.
Основные проявления синдрома – это психическая и умственная неполноценность. Может сочетаться с множественными пороками развития: сердца, спинного и головного мозга, органов мочевыводящей системы и желудочно-кишечного тракта. Так как имеется дефект развития определённых белковых систем, дети с синдромом Дауна меньше ростом и более склонны к развитию ожирения.
Так как часто есть дефект работы иммунной системы, пациенты чаще подвержены тяжёлому течению респираторных болезней. После 20 лет люди с синдромом Дауна начинают стареть быстрее, чем здоровые. К 40 годам у части из них развивается синдром Альцгеймера.
В 92% случаев синдрома Дауна происходит трисомия по 21 хромосоме. Нарушается расхождение этой пары хромосом. И поэтому во всех соматических клетках больного содержится три 21 хромосомы вместо двух.
В 3% случаев возникает мозаичная форма синдрома. Трисомия по 21 хромосоме встречается только в части клеток организма. Чаще всего – в нейронах головного мозга. В остальных клетках будет или нормальный кариотип с двумя 21 хромосомами или одна 21 хромосома. Это называется моносомия по 21 хромосоме. В этом случае все проявления синдрома Дауна не так ярко выражены. Иногда диагностируются лишь во взрослом возрасте.
В 5% случаев будет транслокационная форма синдрома. 21 хромосома целиком или определёнными частями сцепляется с другой хромосомой. Чаще всего – с 15.
Интеллектуальный дефицит при данной форме выражен слабо, зато наследственность более неблагоприятная. Риск рождения ребёнка с синдромом Дауна в три раза выше, чем при трисомии по 21 хромосоме.
Частота заболевания – 1 на 700 живых новорождённых. Дети, которые погибли внутриутробно, в эту статистику не входят. Чем младше мать, тем меньше риски развития такой аномалии. У матерей старше 45 лет риск составляет уже 1 на 30.
Самый точны й метод диагностики – это исследование хромосом плода, которые попали в околоплодные воды матери или содержатся в оболочке плода – хорионе. Процедура забора околоплодных вод называется амниоцентезом, а хориона – хорионобиопсией. Её проводят только по показаниям. После обнаружения косвенных признаков синдрома Дауна на УЗИ и патологии уровня биохимических показателей: свободного эстриола, альфа-фетопротеина и хорионического гонадотропина.
Специфического лечения синдрома Дауна нет.
Есть и другие патологии.
- Синдром Патау. Синдром трисомии по 13 хромосоме. Проявляется недоразвитием нервной системы, расщелиной твёрдого и мягкого нёба, полидактилией, увеличением почек. В 80% случаев встречаются пороки сердца. Тяжёлые нарушения в работе нервной системы приводят к тому, что на первом году жизни умирает примерно 95% таких детей.
- Синдром Эдвардса. Трисомия по 18 хромосоме. Проявляется деформациями костей черепа, пороками развития сердца и лёгких. Характерно недоразвитие мозжечка и умственная отсталость. Внутриутробно дети значительно отстают в весе и росте. Как и при синдроме Патау, только 5% детей доживают до возраста 1 года.
- Синдром Шерешевского – Тернера. Хромосомное заболевание, при котором у женщин нет одной Х-хромосомы. Или же она дефектна. Кариотип таких больных может быть 45 Х0, 45 Х/ 46 ХХ, 45 Х/ 46 ХY. Для патологии характерны низкорослость, непропорциональное туловище и конечности, первичное бесплодие, ожирение. Визуально могут быть крыловидные складки кожи в области шеи. Интеллект чаще всего сохранён.
- Синдром Клайнфельтера. Дополнительные Y или X-хромосомы у мужчин. На каждые 500 новорождённых мальчиков приходится один с таким синдромом. Клинически заметным синдром становится лишь после наступления периода пубертата. Больные имеют высокий рост, развитые грудные железы. Плохо развитые органы репродуктивной системы приводят к первичному бесплодию и эректильной дисфункции.
В России всем новорождённым в родильных домах делают неонатальный скрининг – обследование на тяжёлые генетические болезни, которые никак не проявляются сразу после рождения. При появлении признаков болезни они уже необратимы, но вовремя начатое лечение помогает вырастить максимально здорового ребёнка.
Из пятки малыша берут каплю крови и наносят на специальную фильтровальную бумагу. Бумагу высушивают и передают в городской или районный генетический центр, где проводят исследование.
Раньше определяли лишь 5 наследственных заболеваний. В 2022 году в рамках пилотного проекта к ним присоединилось исследование на спинальную мышечную атрофию. С 1 января 2023 года скрининг расширен до 36 патологий. Это первичные иммунодефициты, болезни обмена веществ, врождённый гипотиреоз, адреногенитальный синдром, муковисцидоз. Все они поддаются терапии.
Важно: статья не является медицинской рекомендацией. Для получения подробной информации обратитесь к специалисту.
Фото: Freepik.com (за исключением указанных особо)

