Причины возникновения
Синдром Марфана — классическое генетическое заболевание. Оно имеет характерные особенности:

- наследование синдрома — аутосомно-доминантное. Больные с нарушениями встречаются в каждом поколении при наличии генетической аномалии у родителей;
- изменения во внутренних органах носят множественный характер и затрагивают различные органы и системы.
Нарушения при синдроме Марфана наблюдаются в гене, кодирующем белок фибриллин. Он является важным компонентом внеклеточного вещества и обеспечивает его эластичность и способность к сокращению. Мутации возникают спонтанно в половых клетках, которые после слияния приводят к образованию зиготы.
Виды заболевания

У детей выделяют несколько типов синдрома. Выделяют стертый и выраженный варианты заболевания. При стертом варианте синдрома Марфана клинические проявления выражены слабо и затрагивают одну-две системы внутренних органов. Дети с подобным течением патологии могут долгое время не иметь диагноза, который ставится после генетического исследования. При выраженном варианте болезни имеются симптомы поражения трех и более систем организма, причем в одной из них признаки выражены явно.
В зависимости от течения синдрома выделяются прогрессирующий и стабильный тип патологии. Стабильный вариант характеризуется отсутствием развития симптомов на протяжении многих лет. В отличие от него, при прогрессирующей болезни проявления заболевания прогрессируют.
Клинические проявления
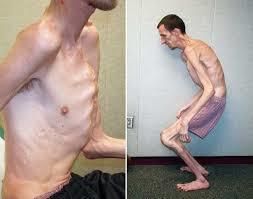
При синдроме Марфана характерно поражение сердечно-сосудистой системы, опорно-двигательного аппарата и глаз. Патология характеризуется множественными признаками, которые различаются у отдельных больных. Синдром имеет хроническое течение, однако скорость прогрессирования симптомов индивидуальна.
Пациенты с заболеванием отличаются высоким ростом. Длина конечностей больше, чем размер туловища (долихостенмелия). Характерный симптом — длинные паукообразные пальцы (арахнодактилия). Телосложение астеническое с невыраженной подкожной жировой клетчаткой и слабо развитыми скелетными мышцами. Лицо узкое и высокое. Возможны нарушения прикуса и аркообразное небо. Синдром Марфана можно заподозрить у новорожденного. Длина тела после рождения у мальчиков обычно больше 53 см, а у девочек — 52,5 см.
При подозрениях на синдром Марфана необходимо проконсультироваться с врачом-генетиком. Специалист проведет внешний осмотр ребенка и при необходимости назначит дополнительные методы исследования.
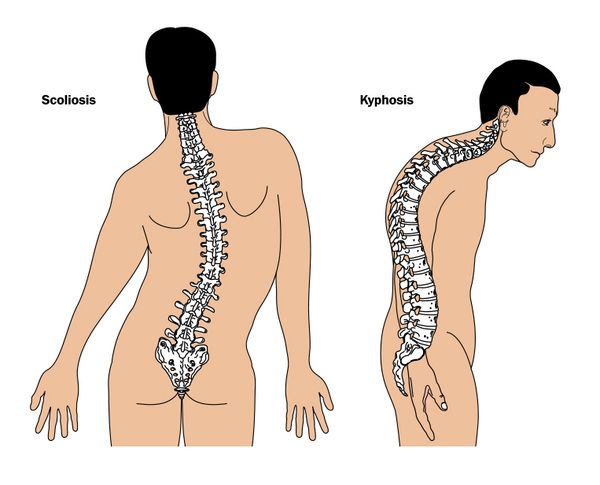
К признакам синдрома относят гипермобильность суставов. Это состояние, при котором человек может выполнять движения в суставных сочленениях в большем диапазоне, чем здоровые люди. Изменения в соединительной ткани приводят к деформациям грудной клетки, искривлениям позвоночника, плоскостопию и другим ортопедическим нарушениям.
Клиническое течение и исход заболевания зависят от выраженности сердечно-сосудистых изменений. У пациентов наблюдаются дефекты в эластических сосудах (аорта, крупные артерии), пороки клапанов и перегородок сердца. Аорта имеет расширения в начальных отделах, что нарушает работу аортального клапана в левом желудочке сердца, а также множественные аневризмы. Врожденные пороки сердца различны: стеноз легочной артерии, дефекты межжелудочковой и межпредсердной перегородки. Для детей с заболеванием характерны изменения ритма различной степени тяжести.
Патология сердечно-сосудистой системы — основной фактор, влияющий на продолжительность жизни. Врожденные пороки при наличии показаний нуждаются в хирургическом устранении.
Нарушения зрения встречаются у большинства больных. Чаще всего выявляются близорукость, изменения положения хрусталика, нарушения строения роговицы, радужной оболочки, расходящееся косоглазие и др. Зрение у детей прогрессивно ухудшается и требует коррекции.
Помимо указанных систем, страдают и другие внутренние органы. Патологические изменения наблюдаются в головном и спинном мозге, легких, кожном покрове, венах, почках и др. Все они возникают из-за нарушений в строении соединительной ткани, которая необходима для поддержания целостности и эластичности структур в органах.
Диагностические манипуляции
Диагностика синдрома основывается на изучении семейного анамнеза, физикальном осмотре и данных, полученных при проведении ЭКГ, ЭхоКГ, рентгенографических методов, лабораторных и молекулярно-генетических исследованиях. Имеются критерии для постановки диагноза:
- изменения в восходящем отделе аорты в виде ее расширения или расслоения;
- патологическое расположение хрусталика;
- деформация грудины, являющаяся показанием для оперативного лечения;
- верхний сегмент тела по длине превышает нижний;
- выраженный сколиоз;
- ограничение амплитуды движений в колене и локте.
На ЭКГ выявляются нарушения сердечного ритма и гипертрофические изменения в миокарде. ЭхоКГ (ультразвуковое исследование сердца) позволяет обнаружить недостаточность клапанов, обратный ток крови при сокращениях миокарда и другие нарушения строения. Гипертрофия сердца и изменения в аорте диагностируются при рентгенографии, компьютерной или магнитно-резонансной томографии органов грудной клетки.
Все пациенты с синдромом Марфана нуждаются в консультации с офтальмологом. Специалист проводит биомикроскопию и офтальмоскопию для оценки положения хрусталика. Дополнительно назначают и другие методы: рентгенографию крупных суставов, МРТ головного и спинного мозга и др.
Дифференциальная диагностика основывается на исключении других генетических и хромосомных заболеваний. К ним относят гомоцистинурию, наследственную артроофтальмопатию, синдром Элерса – Данлоса и пр.
Подходы к лечению
В терапии заболевания участвуют несколько специалистов: генетик, терапевт, офтальмолог, ортопед, кардиолог и кардиохирург. Это необходимо в связи с комплексными патологическими изменениями во внутренних органах. Основная задача лечения — предупредить прогрессирование синдрома и развитие его осложнений.
Оперативные вмешательства проводятся при выраженной недостаточности клапанного аппарата сердца, расслоении или значительном расширении восходящего отдела аорты. Операции с реконструкцией аорты позволяют в 2–3 раза увеличить ожидаемую продолжительность жизни пациента. При стенозе или недостаточности клапанов показано проведение их протезирования. Дополнительно используют бета-адреноблокаторы, снижающие риск сердечных осложнений.
Больным необходимо соблюдать назначения лечащего врача. Попытки самолечения, включающие в себя изменения дозировок и схем приема лекарств, приводят к прогрессированию болезни.
Коррекция зрения проводится с помощью контактных линз или очков. Если у пациента развилась катаракта или глаукома, то возможно оперативное лечение заболеваний. При прогрессировании смещения хрусталика его заменяют на искусственный.
Ортопедическая патология с выраженными изменениями в опорно-двигательном аппарате устраняется при помощи хирургических вмешательств: эндопротезирования, торакопластики, стабилизации позвоночника и др. Помимо указанных операций, пациенту проводится послеоперационная антибиотикотерапия, назначаются витамины и препараты, улучшающие метаболизм.
Пациенты с диагностированным синдромом Марфана нуждаются в постоянном наблюдении у врача. Каждый год проводится комплексное обследование, направленное на оценку состояния систем внутренних органов.
Прогноз при стертой форме или раннем выявлении заболевания положительный. Своевременное устранение пороков развития сердечных клапанов и аорты позволяет увеличить продолжительность жизни пациента. В тех случаях, когда человек долгое время не обращался за медицинской помощью, возможно развитие тяжелых осложнений с сердечной недостаточностью.
Видео

Читайте далее: синдром морриса